
December 06, 2024
December 06, 2024

November 11, 2024

November 11, 2024

November 11, 2024

October 10, 2024
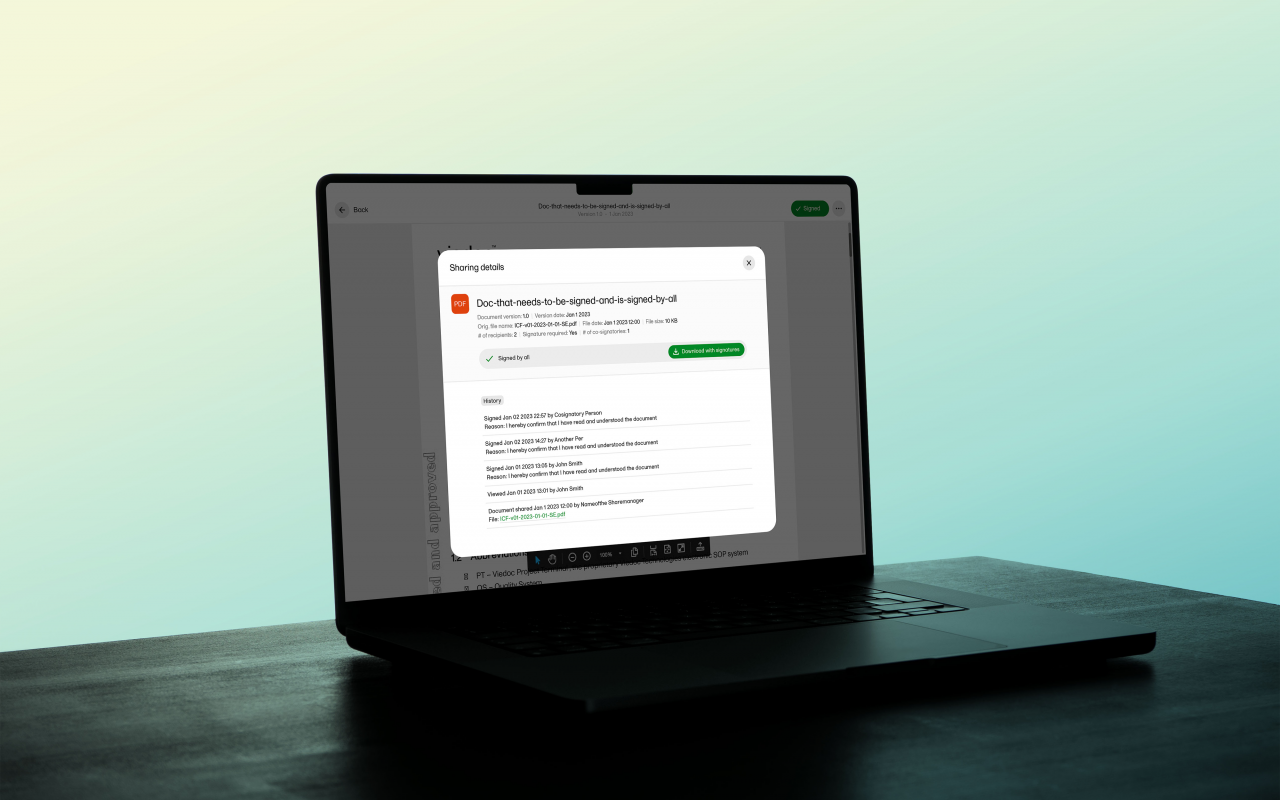
October 09, 2024

September 04, 2024
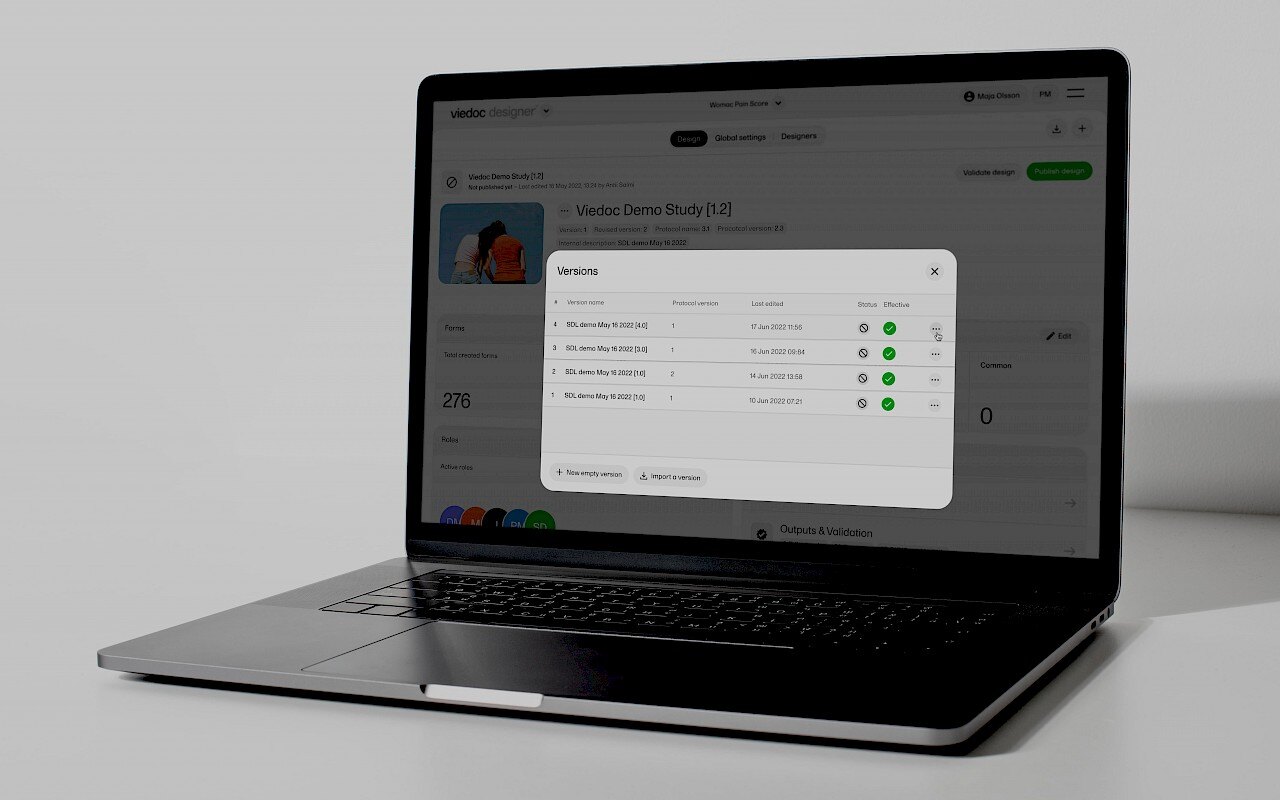
August 22, 2024

July 30, 2024

July 16, 2024

June 27, 2024