
April 17, 2025
April 17, 2025
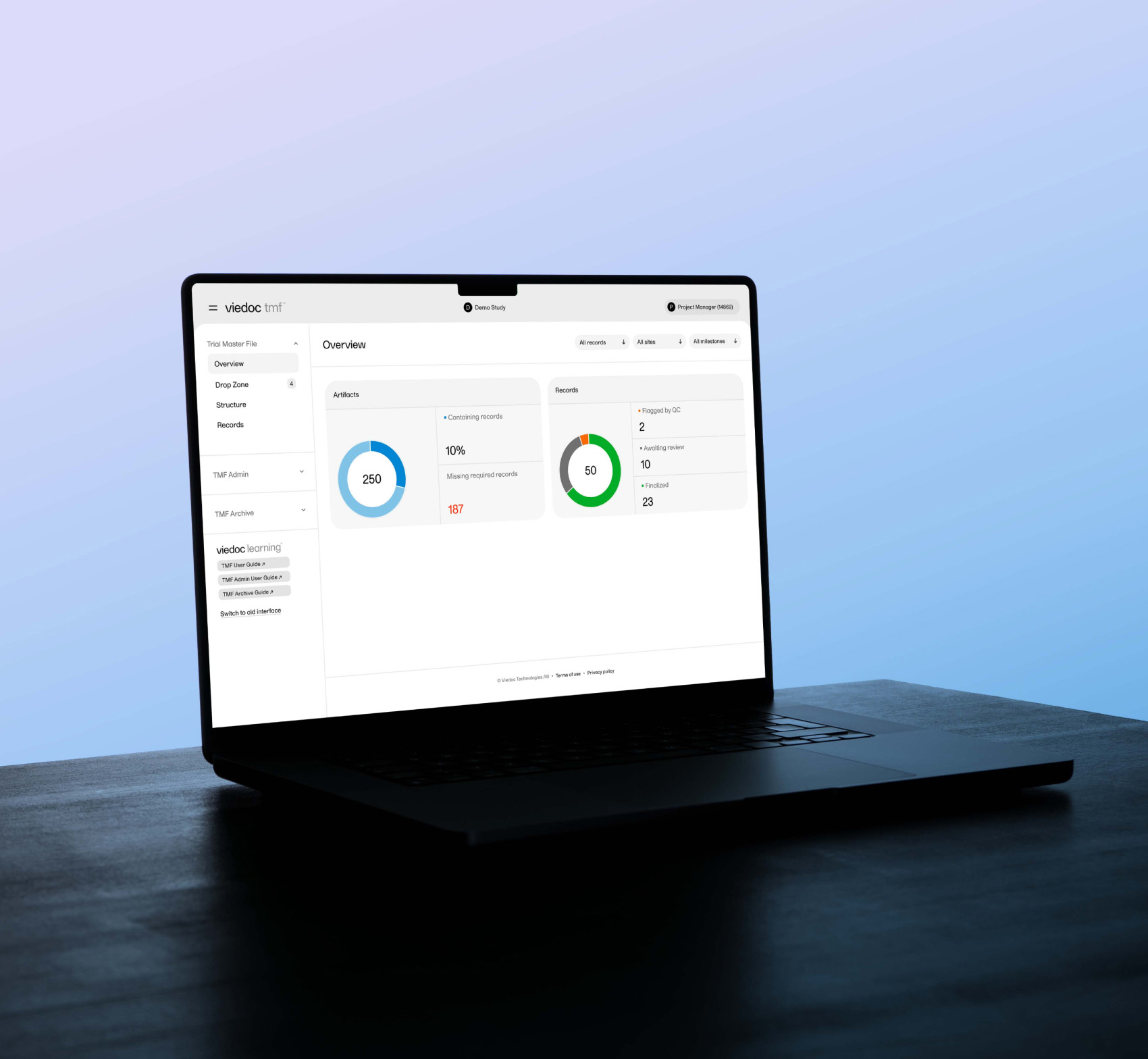
March 31, 2025

March 17, 2025

March 13, 2025

February 20, 2025
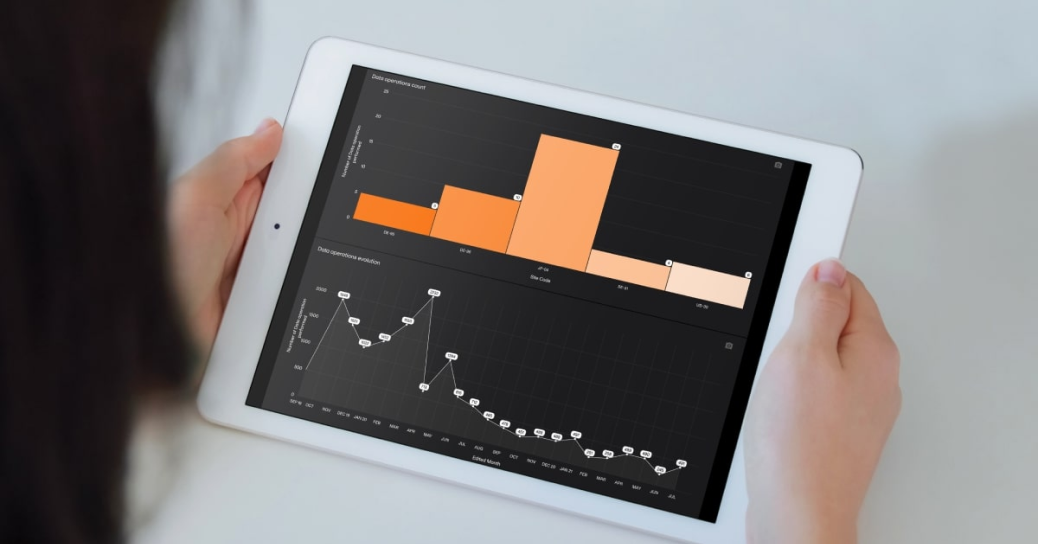
February 03, 2025

January 20, 2025

January 09, 2025

December 06, 2024

November 11, 2024

November 11, 2024